نوروفیبروماتوز چیست؟
نوروفیبروماتوز
نوروفیبروماتوز یک اختلال ژنتیکی است که سبب ایجاد تومور در بافت عصبی میشود. این تومورها میتوانند در هر نقطه از سیستم عصبی، از جمله مغز، نخاع و اعصاب ایجاد شوند. نوروفیبروماتوز معمولا در دوران کودکی یا اوایل بزرگسالی تشخیص داده میشود. تومورها غیرسرطانی (خوش خیم) هستند، اما گاهی اوقات میتوانند سرطانی (بدخیم) شوند. نشانهها اغلب خفیف هستند. با این حال، عوارض نوروفیبروماتوز میتواند شامل کاهش شنوایی، اختلال یادگیری، مشکلات قلبی و رگهای خونی (قلب و عروق)، از دست دادن بینایی و درد شدید باشد.
درمان نوروفیبروماتوز با هدف به حداکثر رساندن رشد و پیشرفت سالم و مدیریت عوارض به محض ایجاد انجام میشود. هنگامی که نوروفیبروماتوز باعث ایجاد تومورهای بزرگ یا تومورهایی میشود که بر روی یک عصب فشار میآورند، جراحی میتواند به کاهش علائم کمک کند. بعضی از افراد ممکن است از سایر روشهای درمان، مانند رادیو تراپی استریو تاکتیک یا داروهای کنترل درد استفاده کنند.
انواع
دو نوع نوروفیبروماتوز وجود دارد که هر کدام دارای علایم و نشانههای مختلف هستند.
نوروفیبروماتوز نوع یک
نوروفیبروماتوز 1 (NF1) معمولا در دوران کودکی ظاهر میشود. نشانهها اغلب در هنگام تولد و یا کمی بعد، و تقریبا همیشه تا سن 10 سالگی آشکار هستند. علائم و نشانهها اغلب خفیف تا متوسط هستند اما میتوانند در شدت متفاوت باشند.
نشانهها و علائم عبارتند از:
- لکههای صاف قوهای روشن بر روی پوست (لکههای شیرقهوه):این لکههای بیضرر در بسیاری از افراد رایج هستند. داشتن بیش از شش لکه شیر قهوه یک نشانه قوی از نوروفیبروماتوز 1 است. آنها معمولا در هنگام تولد یا در اولین سالهای زندگی ظاهر میشوند و پس از آن تثبیت میشوند.
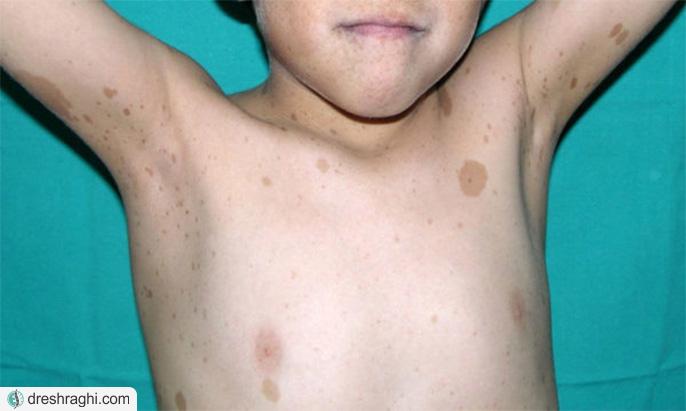
- کک و مکها در ناحیه زیر بغل یا کشاله ران:کک و مکها معمولا در سن 3 تا 5 سالگی ظاهر میشود. کک و مکها کوچکتر از شش لکه شیرقهوه هستند و در چینهای پوست به صورت دستهای ایجاد میشوند.
- برآمدگیهای روی عنبیه چشم (ندولهای لیش):این ندولهای بیضرر را نمیتوان به آسانی مشاهده کرد و بینایی شما را تحت تاثیر قرار نمیدهند.
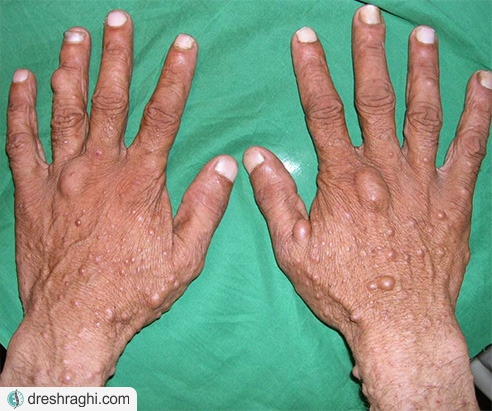
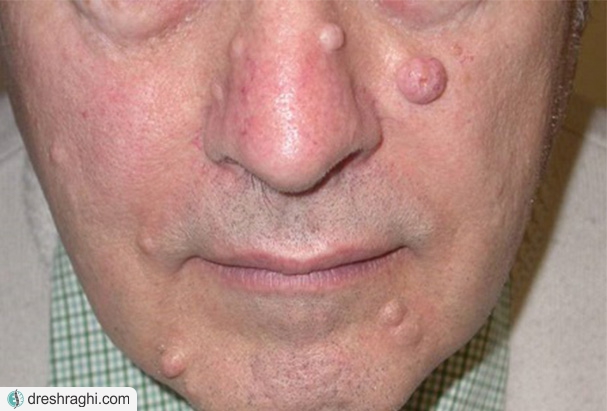
- برآمدگیهای نرم رو یا زیر پوست (نوروفیبروما):این تومورهای خوش خیم معمولا در زیر و یا روی پوست ایجاد میشوند، اما همچنین میتوانند در داخل بدن رشد کنند. گاهی اوقات، رشد شامل اعصاب متعدد (نوروفیبروما شبیه شبکه) میشود.
- تغییر شکل استخوان: رشد غیر طبیعی و کمبود تراکم معدنی استخوان میتواند باعث بدشکلی استخوان مانند ستون فقرات منحنی یا قوز (اسکلیوز) و یا پای پرانتزی شود.
- تومور در عصب بینایی)گلیومای چشمی(: این تومورها معمولا در سن 3 سالگی و به ندرت در اواخر دوران کودکی و نوجوانی ظاهر میشوند، و تقریبا هرگز در بزرگسالان ظاهر نمیشود.
- ناتوانیهای یادگیری:مهارتهای تفکر معیوب در کودکان مبتلا به نروفیبروماتوز نوع 1 رایج هستند، اما معمولا خفیف میباشند. اغلب معلولیت یادگیری خاص مانند مشکل با خواندن یا ریاضیات وجود دارد. اختلال پیش فعالی یا کمبود توجه (ADHD) نیز رایج هستند.
- اندازه سر بزرگتر از حد متوسط:کودکان مبتلا به نروفیبروماتوز نوع 1 به علت افزایش حجم مغز، اندازه سر بزرگتر از حد معمول دارند.
- قد کوتاه: قد کودکان مبتلا به نروفیبروماتوز نوع 1 کمتر از حد وسط است.
نروفیبروماتوز نوع دو
نوروفیبروماتوز 2 (NF2) بسیار کمتر از نروفیبروماتوز نوع 1 رایج است. علائم و نشانههای نروفیبروماتوز نوع 2 معمولا ناشی از رشد تومورهای خوش خیم و رشد آهسته (آکوستیک نوروما) در هر دو گوش است. این تومورها، که با عنوان شوآنوم وستیبولار نیز شناخته میشوند، روی عصبی رشد میکنند که اطلاعات صوت و تعادل را از داخل گوش به مغز حمل میکنند.
نشانهها و علائم معمولا در آخرای نوجوانی و سالهای اولیه بزرگسالی ظاهر میشوند و میتوانند در شدت متفاوت باشد. علائم و نشانهها شامل موارد زیر هستند:
- از دست دادن شنوایی تدریجی
- زنگ زدن در گوشها
- تعادل ضعیف
- سر درد
گاهی اوقات نوروفیبروماتوز نوع 2 میتوانند منجر به رشد شوانوما در اعصاب دیگر بدن، از جمله اعصاب جمجمعهای، نخاعی، و اعصاب بینایی و محیطی شود. علائم و نشانههای این شوانوما میتوانند شامل موارد زیر باشند:
- بیحسی و ضعف در دستان و پاها
- درد
- مشکلات تعادل
- افتادگی صورت
- مشکلات دید و یا مبتلا شدن به آب مروارید
علت ایجاد
نوروفیبروماتوز توسط نقایص ژنتیکی (جهش) بوجود میآید که یا توسط یکی از والدین به ارث برده میشوند یا به صورت خودبخودی در نطفه بسته شده رخ میدهند. ژنهای خاصی که درگیر هستند، به نوع نوروفیبروماتوز بستگی دارند:
- نروفیبروماتوز نوع 1:ژن نروفیبروماتوز نوع 1 بر روی کروموزوم 17 قرار دارد. این ژن به طور معمول یک پروتئین به نام نوروفیبرومین تولید میکند که به تنظیم رشد سلول کمک میکند. ژن جهش یافته باعث از بین رفتن نوروفیبرومین میشود که اجازه میدهد سلولها بدون کنترل رشد کنند.
- نروفیبروماتوز نوع 2:ژن نروفیبروماتوز نوع 2 بر روی کروموزوم 22 قرار دارد و یک پروتئین با نام مرلین تولید میکند. ژن جهش یافته منجر به از دست رفتن مرلین شده، که منجر به رشد کنترل نشده سلولها میشود.
عوارض
عوارض نوروفیبروماتوز حتی در یک خانواده یکسان نیز متفاوت است. به طور کلی، عوارض ناشی از رشد بافت عصبی تحریک کننده تومور یا فشار بر اندامهای داخلی هستند.
عوارض نروفیبروماتوز نوع 1 عبارتند از:
- مشکلات عصبی:مشکلات یادگیری و فکری رایجترین مشکلات عصبی مرتبط با نروفیبروماتوز نوع 1 هستند. عوارض غیر متداول عبارتند از: صرع و ایجاد مایع اضافی در مغز.
- نگرانیهای مربوط به ظاهر: علائم قابل مشاهدهی نوروفیبروماتوز، مانند لکههای شیر قهوه بزرگ، نوروفیبروماهای متعدد در ناحیه صورت یا نوروفیبروماهای بزرگ، میتوانند ناشی از اضطراب و اندوه احساسی باشند، حتی اگر از نظر پزشکی جدی نباشند.
- مشکلات اسکلتی:بعضی از کودکان دارای استخوانهای شکل گرفته به طور غیر طبیعی هستند که میتواند باعث خمیدگی پاها و شکستگیها شود که گاهی اوقات بهبود نمییابند. نروفیبروماتوز نوع 1 میتواند انحنای ستون فقرات (اسکلیوز) را ایجاد کند که ممکن است نیاز به جراحی داشته باشد. نروفیبروماتوز نوع 1 همچنین با کاهش تراکم معدنی استخوان همراه است که خطر ابتلا به ضعف استخوان را افزایش میدهد (پوکی استخوان).
- مشکلات دید:گاهی اوقات در کودکان، گلیومای چشمی میتواند ایجاد شود، که بر روی دید چشم تاثیر میگذارد.
- مشکلات تغییر هورمونی در طول زمان:تغییرات هورمونی مربوط به بلوغ، بارداری یا یائسگی ممکن است سبب افزایش نوروفیبروم شوند. اکثر زنان مبتلا به نروفیبروماتوز نوع 1 دارای بارداری سالم هستند، اما احتمالا نیاز به مراقبت توسط یک متخصص زنان و زایمان آشنا به عارضه دارند.
- مشکلات قلبی عروقی:افراد مبتلا به نروفیبروماتوز نوع 1 دارای افزایش خطر ابتلا به فشار خون بالا و به ندرت بیقاعدگیهای رگهای خونی هستند.
- مشکلات تنفس:به ندرت، نوروفیبروم پلکسی فورم ممکن است روی راه هوایی شما فشار وارد کند.
- سرطان: حدود 3 تا 5 درصد از افراد مبتلا به نروفیبروماتوز نوع 1 مبتلا به تومورهای سرطانی میشوند. اینها معمولا از نوروفیبرومهای زیر پوست یا از نوروفیبروم پلکسی فورم ایجاد میشوند. افراد مبتلا به نروفیبروماتوز نوع 1 همچنین دارای خطر بیشتری نسبت به انواع دیگر سرطان، مانند سرطان پستان، سرطان خون، تومورهای مغزی و برخی انواع سرطان بافت نرم هستند.
- تومور خوش خیم غده فوق کلیه (فئوکروموسیتوما):این تومور غیر سرطانی هورمونهایی را ترشح میکند که فشار خون شما را افزایش میدهند. فئوکروموسیتوما به طور کلی با جراحی حذف میشود.
عوارض نروفیبروماتوز نوع 2 عبارتند از:
- ناشنوایی کلی یا جزئی
- آسیب عصبی صورت
- مشکلات دید
- تومورهای پوستی خوش خیم کوچک (شوانوما پوست)
- ضعف یا بیحسی در اندامها
- تومورهای چندگانه خوش خیم مغز یا تومورهای ستون فقرات که نیاز به جراحیهای مکرر دارند (مننژیوما)
تشخیص
پزشک شما با معاینه فیزیکی و بررسی سابقه پزشکی شما و سابقه پزشکی خانواده شروع خواهد کرد. نروفیبروماتوز نوع 1 اغلب میتواند بر اساس معاینه فیزیکی تشخیص داده شود. پزشک شما ممکن است از یک لامپ ویژه برای بررسی پوست شما برای لکههای شیرقهوه استفاده کند. معاینه فیزیکی و سابقه خانوادگی نیز برای تشخیص نروفیبروماتوز نوع 2 مهم هستند.
پزشک شما همچنین ممکن است موارد زیر را توصیه کند:
- معاینه چشم:چشم پزشک میتواند برجستگیهای رنگی در عنبیه چشم (نودل های لیش) و آب مروارید را تشخیص دهد.
- معاینه گوش: آزمایشاتی مانند سنجش شنوایی، الکترونیستاگموگرافی، و پتانسیل برانگیخته شنوایی ساقه مغز میتوانند به بررسی مشکلات شنوایی و تعادل در افراد مبتلا به نروفیبروماتوز نوع 2 کمک کنند.
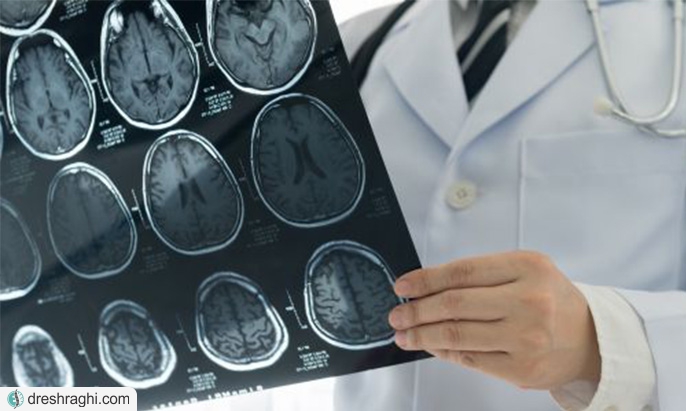
- آزمایشات تصویربرداری: اشعه ایکس، سی تی اسکن یا ام آر آی (لینک به ام آر آی و سی تی اسکن) میتوانند به شناسایی ناهنجاریهای استخوانی، تومورهای مغز یا طناب نخاعی و تومورهای بسیار کوچک کمک کنند. ممکن است ام آر آی برای کمک به شناسایی گلیوماهای چشمی استفاده شوند. آزمایشات تصویربرداری همچنین اغلب برای نظارت بر نروفیبروماتوز نوع 2 و شوانوماتوزیز استفاده میشوند.
- آزمایشات ژنتیک: آزمایشات برای شناسایی نروفیبروماتوز نوع 1 و نروفیبروماتوز نوع 2 در دسترس هستند و میتوانند قبل از زایمان انجام شوند. از پزشک درباره مشاوره ژنتیکی بپرسید. آزمایشات ژنتیکی برای شوانوماتوزیز محدود هستند.
برای تشخیص نروفیبروماتوز نوع 1، باید حداقل دو نشانه از عارضه را داشته باشید. اگر فرزند شما تنها یک علامت داشته باشد و سابقه خانوادگی نروفیبروماتوز نوع 1 را نداشته باشد، پزشک شما احتمالا کودک را برای ایجاد علائم اضافی نظارت خواهد کرد. تشخیص نروفیبروماتوز نوع 1 معمولا در سن 4 سالگی انجام میشود. آزمایش ژنتیک ممکن است به تعیین تشخیص کمک کند.
درمان
نوروفیبروماتوز نمیتواند علاج شود، اما درمانها برای علائم و نشانههای شما در دسترس هستند. به طور کلی، هر چه زودتر شما یا فرزندتان تحت مراقبت پزشک متخصص در درمان نوروفیبروماتوز قرار بگیرید، نتیجه بهتر است.
پیگیری
اگر فرزند شما مبتلا به نروفیبروماتوز نوع 1 باشد، پزشک احتمالا برای موارد زیر معاینه مناسب سن سالانه را توصیه میکند:
- پوست کودک خود را برای نوروفیبرومهای جدید یا تغییرات در نوروفیبرومهای موجود ارزیابی کنید.
- نشانههای فشار خون بالا را بررسی کنید.
- با توجه به نمودارهای رشد موجود برای کودکان مبتلا به نروفیبروماتوز نوع 1، رشد و پیشرفت کودک خود را ارزیابی کنید، از جمله قد، وزن و اندزه سر.
- نشانههای بلوغ زودرس را بررسی کنید.
- کودک خود را برای هر گونه تغییر اسکلتی و بیقاعدگیها بررسی کنید.
- رشد یادگیری و پیشرفت کودک در مدرسه را ارزیابی کنید.
- یک معاینه کامل چشم انجام دهید.
در صورتی که متوجه هرگونه تغییر در نشانهها و علائم در بین ملاقاتها شدید، بلافاصله با پزشکتان تماس بگیرید. مهم این است که امکان تومور سرطانی را بررسی کنید و در مرحله اولیه درمان مناسب را بدست آورید.
جراحی
پزشک شما ممکن است عمل جراحی یا سایر روشها را برای درمان علائم شدید یا عوارض نوروفیبروماتوز توصیه کند.
- جراحی برای حذف تومورها:علائم میتوانند با از بین بردن تمام یا بخشی از تومورهایی که فشرده سازی بافت اطراف یا ارگانهای آسیب دیده را تحت فشار قرار میدهند، کاهش یابند. اگر مبتلا به نروفیبروماتوز نوع 2 هستید و از دست دادن شنوایی، فشردگی ساقه مغزی یا رشد تومور را تجربه میکنید، پزشک ممکن است عمل جراحی را به منظور حذف نوروم آکوستیک که باعث ایجاد مشکلات شما میشوند، توصیه کند. حذف کامل شوانوما در افراد مبتلا به سوانوماتوزیز میتواند درد را به طور قابل توجهی کاهش دهد.
- رادیو سرجری استریوتاکتیک:این روش پرتو را دقیقا به تومور شما میفرستد و نیازی به برش ندارد. در صورتی که مبتلا به نروفیبروماتوز نوع 2 هستید، رادیو سرجری استریوتاکتیک ممکن است یک گزینه برای حذف نورینوم آکوستیک باشد. رادیو سرجری استریوتاکتیک میتواند به حفظ شنوایی شما کمک کند.
- ایمپلنت شنوایی ساقه مغز و ایمپلنت حلزونی:در صورتی که مبتلا به نروفیبروماتوز نوع 2 و از دست دادن شنوایی هستید، این دستگاهها ممکن است به بهبود شنوایی شما کمک کنند.
درمان سرطان
تومورهای بدخیم و سرطانهای دیگر مرتبط با نوروفیبروماتوز با درمانهای استاندارد سرطان، مانند جراحی، شیمی درمانی و پرتو درمانی درمان میشوند. تشخیص زودرس و درمان مهمترین عوامل ناشی از نتیجه خوب هستند.
داروهای درد
مدیریت درد بخش مهمی از درمان برای شوانوماتوزیز است. پزشک شما ممکن است موارد زیر را توصیه کند:
- گاباپنتین (Neurontin) یا پره گابالین (Lyrica) برای درد عصبی
- داروهای ضد افسردگی 3 حلقهای مانند آمی تریپتیلین
- سروتونین و بازدارندههای بازجذب نوراپینفرین مانند دولوکستین (Cymbalta)
- داروهای صرعی مانند توپیرامات (Topamax) یا کاربامازپین (کرباترول، تگراتول)
نوروفیبروماتوز نوع دو (NF2) یک عارضه وراثتی است که در بیشتر موارد به همراه شوانوما وستیبولار دو طرفه، که به آن نورومای آکوستیک نیز گفته میشود، پدید میآید. این عارضهها تومورهای خوش خیم (غیر سرطانی) هستند که روی اعصاب مسئول تعادل در گوش داخلی تشکیل میشوند. اگرچه این تومورها خوشخیم هستند ولی با این حال میتوانند باعث مشکلاتی در شنوایی و تعادل شخص شوند.
بر خلاف نوروفیبروماتوز نوع یک، علت اصلی این بیماری، نوروفیبروماتوز نیست بلکه معمولا این بیماری در اثر شوانومای عصب شنوایی ایجاد می شود. این بیماری با ایجاد تومور های خوش خیم بر روی شبکه عصبی به ویژه اعصاب شنوایی و نزدیک گوش همراه است.
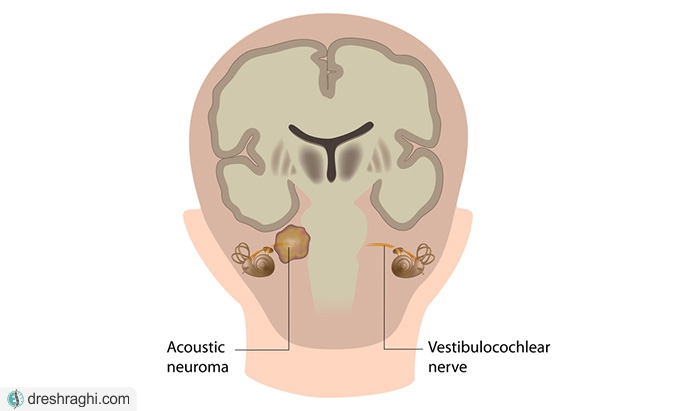
نروفیبروماتوز نوع دو
افراد مبتلا به نوروفیبروماتوز نوع دو بیشتر از سایرین در معرض خطر انواع تومور در سیستم عصبی خود هستند. این تومورها عمدتاً سرطانی نیستند ولی با این حال میتوانند منجر به مشکلات پزشکی قابل توجهی شوند، مخصوصاً اگر چندین تومور داخل یا نزدیک به مغز وجود داشته باشد. سایر انواع تومورهای سیستم عصبی عبارتند از:
- شوانوما در سایر اعصاب
- مننژیوما، یک تومور کُند رشد کننده است که معمولاً روی سطح مغز ایجاد میشود.
- گلیوما، از جمله اپاندیمومادر مغز و طناب نخاعی.
افراد مبتلا به نوروفیبروماتوز نوع دو بیشتر از سایرین در معرض ابتلا به آب مروارید در چشمها و تومورهای پوستی خوش خیم هستند. این ممکن است دارای لکههای قهوهای و سفید باشند، که یک رنگدانه قهوهای رنگ است به رنگ "قهوه با شیر". این ویژگی مشابه کسانی که به نوروفیبروماتوز نوع یک مبتلا هستند، ولی مبتلایان به نوروفیبروماتوز نوع دو معمولاً لکههای قهوهای و سفید کمتری نسبت به مبتلایان به نوروفیبروماتوز نوع یک دارند.
علائم نوروفیبروماتوز نوع دو در اواخر سنین نوجوانی یا اوایل بیست سالگی بروز میکند. ویژگیهای چندگانهای با نوروفیبروماتوز نوع دو مرتبط بوده ولی خطر کلی سرطان پایین است. تعداد ویژگیهای موجود و شدت علائم ممکن است در بین مبتلایان به نوروفیبروماتوز نوع دو متفاوت باشد، حتی اگر هر دو از خانواده باشند.
علت ایجاد
نوروفیبروماتوز نوع دو یک عارضه ژنتیکی است. به این معنا که خطر سرطان و سایر ویژگیهای نوروفیبروماتوز نوع دو میتواند از نسلی به نسل دیگر در یک خانواده انتقال یابد. به ژنهای مرتبط با نوروفیبروماتوز نوع دو (NF2) نیز NF2 گفته میشود. جهش (تغییر) در ژن NF2، که یک "مهار کننده تومور" است، شخص را در معرض خطر فزاینده تومورهای سرطانی و خوش خیم و سایر علائم نوروفیبروماتوز نوع دو (NF2) قرار میدهد. اکثر افراد مبتلا به نوروفیبروماتوز نوع دو (NF2) جهشی در ژن NF2 دارند. تحقیقاتی در حال انجام است تا بیشتر در مورد علل نوروفیبروماتوز نوع دو اطلاعات به دست آوریم.
نحوه وراثت
معمولاً هر سلول 2 نسخه از هر ژن را دارد: یک نسخه که از مادر به ارث میبرد و یک نسخه از پدر. ژن NF2 از یک الگوی ارثی اتوزومال غالب پیروی میکند که در آن جهش ژنتیکی تنها در یک نسخه از ژن پدید میآید. به عبارت دیگر والدین دارای ژن جهش یافته، ممکن است یک نسخه از ژن نرمال خود یا یک نسخه از ژن جهش یافته خود را به کودکشان انتقال دهند. بنابراین، کودکی که پدر یا مادرش دارای ژن جهش یافته باشد، به احتمال 50 درصد جهش ژنتیکی را به ارث خواهد برد. برادر، خواهر، پدر یا مادر شخصی که جهش ژنتیکی دارد نیز 50 درصد احتمال داشتن همان جهش ژنتیکی را خواهد داشت. اگر یک نسخه از ژن جهش یافته به کودک منتقل شود، آن ژن تبدیل به ژن غالب شده و میتواند باعث علائم و ویژگیهای نوروفیبروماتوز نوع یک شود.
گزینههایی برای چنین والدینی که به دنبال بچهدار شدن هستند ولی یکی از والدین ژن جهش یافتهای را دارد که احتمال ابتلا به این سندروم سرطانی وراثتی را افزایش میدهد، وجود دارد. تشخیص ژنتیکی قبل از لانه گزینی (PGD) یک عمل پزشکی است که در ترکیب با لقاح در خارج از بدن یا (IVF) انجام میشود. این روش به کسانی که دارای یک جهش ژنتیکی شناخته شده بخصوص هستند کمک میکند، تا احتمال اینکه بیماری آنها به کودکانشان منتقل شود را کاهش دهند. تخمکهای زن از بدن او گرفته شده و در آزمایشگاه بارور میشود. وقتی که نطفه به یک اندازه مشخصی رسید، یک سلول از او برداشته شده و تحت آزمایش برای وجود این عارضه وراثتی قرار میگیرند. سپس والدین میتوانند تصمیم بگیرند که جنینی را که دارای هیچ جهش ژنتیکی نیست به رحم مادر منتقل کنند. تشخیص ژنتیکی قبل از لانه گزینی (PGD) برای بیش از دو دهه در حال انجام شدن است و در مورد چندین پیش زمینه سندرم وراثتی سرطان انجام گرفته است. هرچند، این فرآیند با در نظر گرفتن عوامل اقتصادی، فیزیکی و احساسی پیش از شروع آن بسیار پیچیده است.
شیوع
تخمین زده میشود که یک نفر از هر 40 هزار نفر به نوروفیبروماتوز نوع دو مبتلا باشد. حدود 50 درصد افراد مبتلا به نوروفیبروماتوز نوع دو هیچ سابقه خانوادگی از این عارضه ندارند. آنها دچار جهش ژنتیکی نوین در ژن NF2 خود هستند.
تشخیص
چندین مجموعه از دستورالعملهای تشخیصی برای تشخیص نوروفیبروماتوز نوع دو پیشنهاد شده و به احتمال زیاد در آینده نیز خواهد شد. این ویژگیها به شدت خاص هستند، و همچنین باید توسط پزشک یا کسی که تجربه درمان نوروفیبروماتوز نوع دو را دارند بررسی شوند تا تشخیص دقیقی صورت گیرد. به طور کلی، کسی که موارد زیر را داشته باشد مشکوک به نوروفیبروماتوز نوع دو خواهد بود:
- شوانوما وستیبولار دو طرفه (در هر دو طرف)، که در واقع تومورهایی هستند که از عصبهایی رشد میکنند که شنوایی و تعادل ما را تحت کنترل دارند.
- پدر یا مادر، برادر یا خواهر یا کودکی مبتلا به نوروفیبروماتوز نوع دو و یکی از موارد زیر:
- شوانوما وستیبولار که پیش از سن 30 سالگی تشخیص داده شده باشد، یا
- دو مورد از این موارد: منینگیوما، گلیوما، شوانوما یا آب مروارید
- شوانوما وستیبولار که پیش از سن 30 سالگی تشخیص داده شود و منینگیوما، گلیوما، شوانوما یا آب مروارید
- چندیدن منینگیوما و شوانوما وستیبولار یک طرفه (تنها در یک طرف) که زیر سن 30 سالگی تشخیص داده شده باشد، گلیوما، شوانوما یا آب مروارید
آزمایش ژنتیک برای بررسی جهش ژنتیکی NF2 برای کسانی که مبتلا به نوروفیبروماتوز نوع دو تشخیص داده شوند وجود دارد.
درمان تومورهای مرتبط با نوع دو
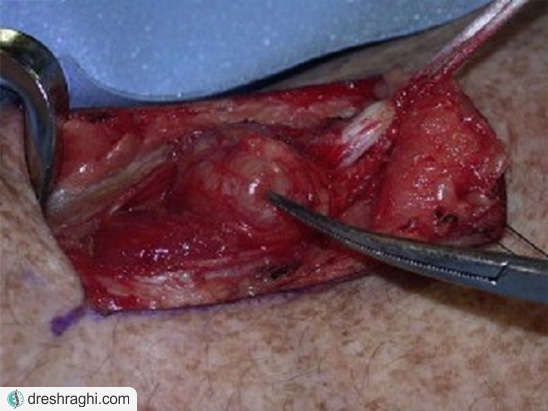
اگرچه روش اصلی درمان تومورهای مرتبط با نوروفیبروماتوز نوع دو تنها جراحی است، ولی بسیاری از افراد مبتلا به این بیماری تومورهایی دارند که یا رشد خیلی کُندی دارد یا اصلاً رشد نمیکند. به عبارت دیگر میتوان این تومورها را با روشی به نام انتظار مراقبتی، مشاهده و انتظار یا مراقبت فعال دقیقاً زیر نظر گرفت. در این روش، درمان فعال هنگامی آغاز میشود که علائمی از بوجود آمدن مشکلات نورولوژیکی یا الگوی رشد تومور عملکرد مغز یا طناب نخاعی را تهدید کند.
مخصوصاً در ارتباط با نورومای آکوستیک دو طرفه، هدف اصلی درمان حفظ قدرت شنوایی شخص تا جای ممکن است. هرچند، نهایتاً قدرت شنوایی بیمار در مرحلهای از بیماری از دست خواهد رفت. جراحی معمولاً برای افرادی انجام میشود که نمیتوانند بشنوند یا در کسانی که تومور کوچکی دارند و میتوان تلاشی معنادار را برای حفظ عصب و همچنین قدرت شنوایی شخص انجام داد.
بعلاوه، پرتودرمانی متمرکز نیز گاهی برای شوانوما وستیبولار مورد استفاده قرار میگیرد. همچون جراحی، این درمان نیز خطر آسیب رسیدن به قدرت شنوایی شخص در تلاش برای کنترل تومور را به همراه دارد. سایر تومورهایی که در این بیماری دیده میشود معمولاً منینگیوما و اپاندیموما هستند، و این نوع تومورها معمولاً تنها در صورتی برداشته میشوند که تا حدی رشد کنند که فشار کافی را روی بافت مغز یا طناب نخاعی پیرامون خود آورده و بر عملکرد آنها تأثیر بگذارند.
گزارشاتی از استفاده از آواستین یا بواسیزوماب، دارویی که با تشکیل عروق خونی تداخل میکند تا مانع از رشد تومور شود، برای درمان شوانوما وستیبولار مرتبط با نوروفیبروماتوز نوع دو وجود دارد که نتایج نوید بخشی را نشان میدهد. هرچند تنها بخش کمی از افراد با این دارو درمان میشوند، ولی نتایج شامل کوچک شدن قابل اندازه گیری تومور و بازیابی نسبی قدرت شنوایی در برخی از بیماران میشود. این درمان هنوز در مرحله آزمایشی است و نیاز به آزمایشات بیشتر دارد ولی با این حال نتایج امیدوار کنندهای را از خود نشان داده است که شاید داروهای دیگر نیز بتوانند در درمان این بیماری موثر باشند.
نحوه پیدا کردن بیماران قبل از بروز بیماری
غربالگریهای پیشنهادی برای مبتلایان به نوروفیبروماتوز نوع دو یا در معرض خطر نوروفیبروماتوز نوع دو عبارتند از:
- انجام اسکن MRI سالانه، که از سنین نوجوانی آغاز میشود
- ارزیابیهایی شنوایی، از جمله شنوایی سنجی و تست پاسخ برانگیخته شنوایی ساقه مغز، این تست فعالیت الکتریکی را در حلزون گوش و مسیرهای شنوایی شناسایی میکند
توصیههای غربالگری ممکن است به مرور زمان تغییر کند زیرا فناوریهای جدیدی بوجود خواهد آمد و افراد بیشتری از نوروفیبروماتوز نوع دو مطلع خواهند شد. اهمیت زیادی دارد که با پزشک خود در مورد نتایج مناسب غربالگری صحبت کنید.
نگارش و گردآورنده: رضا رحیمی
منبع: نوراله اشراقی جراح مغز و اعصاب
- لینک منبع
تاریخ: پنجشنبه , 16 آذر 1402 (20:38)
- گزارش تخلف مطلب